Functional annotation is the process of assigning biological meaning to identified gene sequences. It involves predicting the roles of genes, transcripts, and proteins by comparing them to known databases, identifying protein-coding regions, functional domains, motifs, and gene ontology (GO) terms. The tools used for functional annotation can be roughly divided into two categories: Tools that compare the sequence to be annotated with known data, be it in a database, sequence profiles or other models. And tools that predict features of the sequence without additional data. Tools for the first approach are e.g. BLAST or diamond in combination with SwissProt or UniProt to assign gene names, or cmsearch together with the Pfam database for protein domain profiles to identify protein domains. Tools for the second approach are e.g. COILS for the prediction of coiled-coil regions or TMHMM for the prediction of transmembrane helices in protein sequences.
How to evaluate coiled-coil prediction tools
Coiled coils consist of two or more α-helices that wind around each other and give rise to a variety of quaternary supercoil structures. Coiled-coil regions are characterised by hydrophobic residues at the interface between the supercoiled α-helices and by charged and polar amino acids on the outside. Numerous tools have been developed over the last 30 years, such as COILS/NCOILS, PairCoil, MultiCoil, MarCoil, CCHMM and DeepCoil. How would you rate the predictive quality of these tools? Given the hundreds of thousands of protein crystal structures available, the most obvious approach would be to identify coiled-coil regions in the protein structures, apply the coiled-coil prediction tools to the protein sequences of the structures and compare the 3D reality with the prediction. Luckily, there is a tool called SOCKET designed to identify α-helices in coiled-coil arrangement in protein structures. And the named tools were evaluated compared to the SOCKET data (D Simm, K Hatje, S Waack, M Kollmar (2021) Scientific Reports 11, 12439).
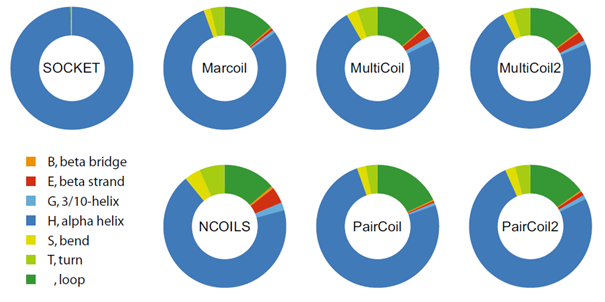
Coiled-coil prediction in protein sequences is completely random
The evaluation results in a binary classification: the predictions either match the coiled-coil regions from the protein structures or they do not. The binary classification leads to four categories: 1) true-positive predictions, i.e. those that match the structures; 2) false-negative regions, i.e. coiled-coil regions of the structures that were not found by predictions; 3) false-positive predictions, i.e. those that do not match the structures; 4) true-negative predictions, i.e. regions without predictions and without coiled-coil regions. Binary categories can be evaluated by the statistical measures such as sensitivity, specificity, accuracy and precision. However, these measures only take two or three of the categories into account. The most reliable performance measure that takes all four categories into account is the Matthews Correlation Coefficient (MCC). Fortunately, the MCC is particularly suitable for analysing unbalanced data sets, i.e. data sets in which one of the categories is strongly overrepresented. In the case of the coiled coil analysis, the overrepresented category is that of true negatives. The detailed analysis of the available coiled-coil prediction tools compared to the protein structures from the PDB showed that the performance of the tools is almost random. In fact, about 25% of the predictions did not even match α-helices. The performance was compared to a naive coin toss model, where for each tool the same proportion of coiled-coil predictions with respect to the total dataset size was used to randomly predict coiled-coils in the entire dataset. The performance of the tools was identical to the coin toss results.
Conclusions
These results show that the predictions of the tools have only limited information value. Coiled-coil predictions are often used to interpret biochemical data and are part of in silico functional genome annotation. However, the results show that these predictions should be treated with great caution and need to be supported and validated by experimental evidence. These tools should not be used in genome annotation as the random predictions lead to massive false functional annotations.
Do it right, get your customized genome annotation
Genome annotation is the beginning of biological analyses and the basis for many costly experiments. Start your research with the best data you can get and let us annotate the genome for you.