Biologists are often surprised when protein sequences do not start with methionine because, in nearly all organisms, protein synthesis begins with the codon AUG, which codes for methionine. This is thought to be a fundamental rule of translation: the ribosome recognizes AUG as the start codon, and initiator tRNA brings methionine to begin the polypeptide chain. Therefore, methionine is expected as the first amino acid in any newly synthesized protein. When a mature protein lacks methionine at the start, it suggests post-translational modification — such as removal by specific enzymes — or non-canonical translation initiation, both of which raise interesting biological questions and warrant further investigation. There may also be technical reasons for missing start methionines in genome annotation datasets.
Technical reasons
1) Assembly problems resulting in fragmentation
Genome assemblies are often incomplete and fragmented due to several technical and biological challenges.
Some key reasons for technical difficulties are
A) DNA quality and extraction bias
Degraded DNA or biased extraction methods can affect the quality of sequencing reads, resulting in incomplete assemblies.
B) Read length limitations
Short-read sequencing technologies (e.g., Illumina) produce reads that are too short to span repetitive regions or structural variants, leading to fragmented assemblies.
C) Sequencing errors
Errors in base-calling, especially in long-read sequencing (e.g., Pacific Biosciences, Oxford Nanopore), can lead to assembly mistakes. High-error rates during the sequencing process can complicate the assembly process and reduce accuracy.
D) Low coverage or uneven coverage
Insufficient sequencing depth or uneven coverage across the genome can leave gaps in the assembly or create regions with low confidence.
E) Assembly algorithms
Limitations in assembly software (e.g., incorrect scaffolding, misassembly of repeat regions) can result in incomplete or erroneous assemblies.
F) Contamination
Presence of contaminant DNA (e.g., from other organisms, environmental sources) can interfere with assembly and produce misleading results.
Reasons for biological challenges leading to assembly errors are
A) Repetitive sequences
Highly repetitive regions (e.g., transposable elements, tandem repeats, centromeres, telomeres) are difficult to assemble accurately because short reads cannot distinguish between identical sequences.
B) Structural variations
Large insertions, deletions, inversions, or duplications are hard to detect and assemble correctly, particularly with short-read technologies.
C) Heterozygosity
High levels of heterozygosity in diploid or polyploid organisms can make assembly difficult, leading to fragmented or mixed contigs.
These technical and biological challenges are the reason why genes can often only be partially annotated at the scaffold boundaries (Figure). In these partial genes, either the 5‘ or the 3’ end of the gene or both ends are missing. The incompleteness often also affects the edges of the so-called chromosome-scale scaffolds. The problem can only be solved by creating telomere-to-telomere assemblies, which are currently only available for a limited number of genomes.
2) Assembly problems within scaffolds
Scaffold assembly problems, especially tandem duplication of regions and missing sequences, are usually the result of limited long-read sequence data and limitations in the assembly software. Most commonly, these problems affect DNA sequence repeat regions, but sometimes tandem gene regions are also affected, both protein coding and non-coding (e.g. RNA) regions.
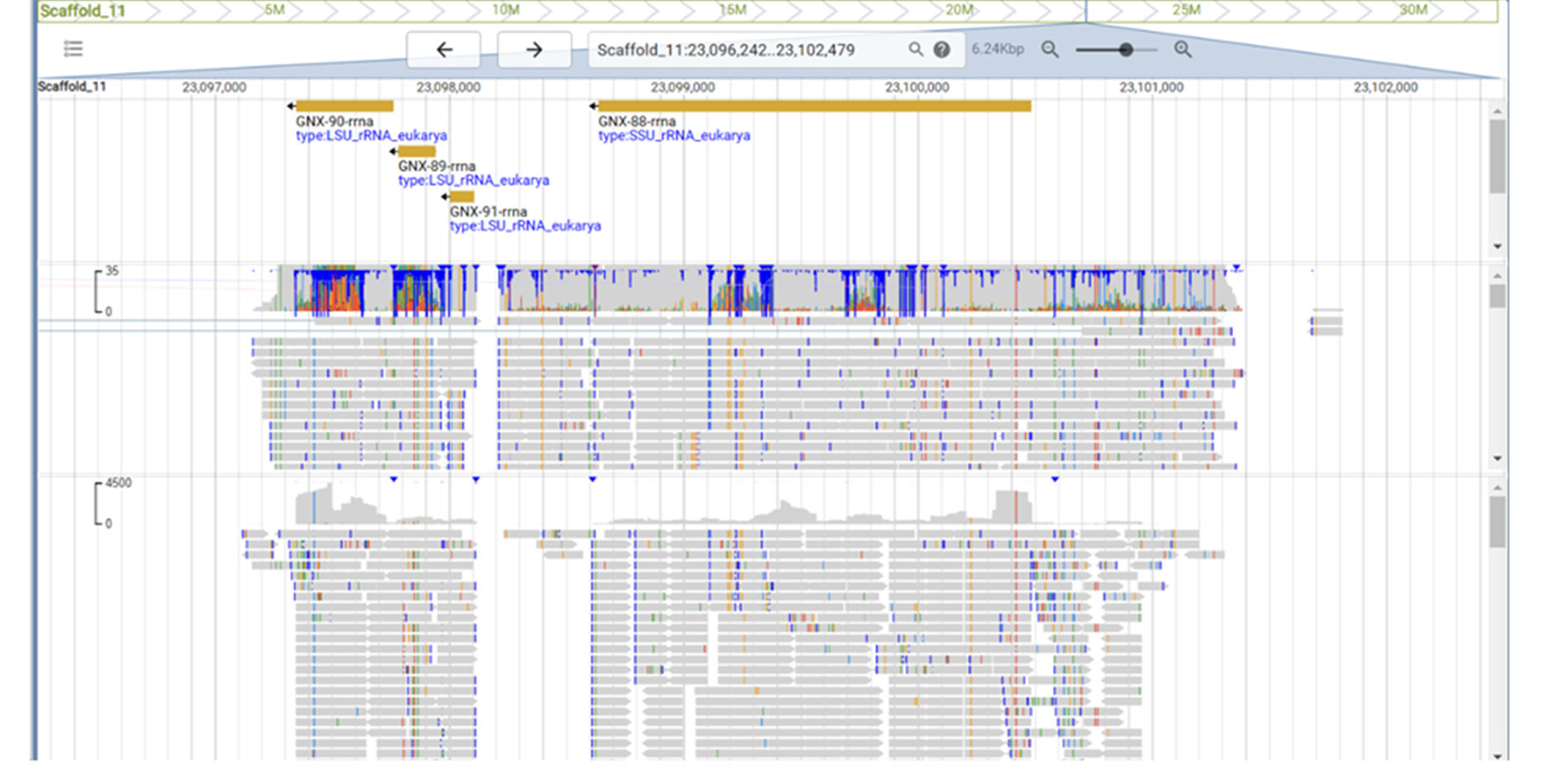
Figure legend: Region around the ribosomal DNA operon in a eukaryote. Eukaryotic RNA genes are organised in a cluster of the 18S RNA (SSU, approx. 2,000 nt long), the 5.8S RNA (150 nt) and the 28S RNA (LSU, 3,000-5,000 nt). The SSU is present here, but only nucleotides `2600 to 3400 of the expected 3400 bp are present from the LSU. This is the only region of the ribosomal DNA operon in this 850 Mbp genome, indicating a problem with internal genome assembly.
Biological reasons
Translation not starting with methionine (ATG)
Although it has been assumed for many decades that translation starts with the methionine codon ATG, ribosome profiling data have shown that up to 50 % of translation starts at non-AUG codons. This figure should not be misunderstood to mean that most cellular proteins originate from non-AUG translation events or that initiation at non-AUG start codons is more efficient than at standard AUG codons. Rather, the data merely indicate that when all initiation sites in the entire transcriptome are counted - without considering the efficiency of individual events - translation initiation occurs more frequently at non-AUG start codons than at AUG codons. Accordingly, when annotating protein-coding genes and transcripts, the start codons for translation and the first coding sequence features (CDS region) are often misinterpreted and therefore incorrectly assigned. The assignment of all start codons to ATG is a purely computer-induced bias.
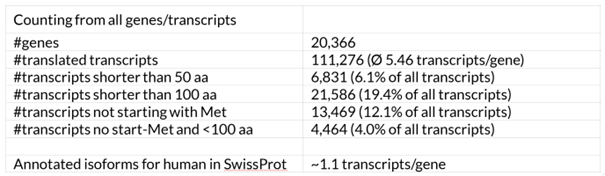
Proteins in the human genome annotation without starting methionine
Researchers normally expect translated transcripts (CDS sequences) to start with a methionine. This is not even true for the official human reference annotation (Table 1). 12.1% of annotated human protein sequences do not start with a methionine, and more than two-thirds of these are longer than one hundred amino acids. Although only 589 of the longest transcripts per gene (2.9 %) are shorter than 100 amino acids, about 20 % of the annotated protein sequences (including alternative isoforms) are shorter than this number. This indicates that a very large proportion of the annotated alternative transcripts are only short pieces compared to their respective longest isoform. It is doubtful that these short isoforms represent functional proteins and not function-restricting isoforms that are never translated.