The BUSCO completeness check assesses the completeness and quality of a genome, transcriptome or proteome assembly by searching for highly conserved single-copy orthologues that should be present in a given lineage. By providing datasets for specific taxonomic lineages, BUSCO helps assess evolutionary completeness. In Part 2, I discussed some differences between the BUSCO completeness check of a genome assembly and the BUSCO completeness check of the genome annotation of that assembly. In Part 3, I will show you another strange case in the BUSCO data that affects completeness.
Example gene: artificially fused gene
BUSCO uses data from OrthoDB for its comparison. The OrthoDB data is derived from the annotations available in GenBank. If the annotations in GenBank are incorrect, the OrthoDB datasets will be affected, and thus the BUSCO datasets will also be affected. In Part 2, I showed an example of a randomly selected gene where six of the ten genes in the gene set contained significant annotation errors. The following example shows two genes that are mostly separated in some BUSCO plant datasets, but were artificially merged in the BUSCO Poales dataset. The two genes are not related at all: one is an S-adenosyl-L-methionine-dependent tRNA-4-demethylwyosine synthase, the other is a methyltransferase. In the BUSCO Solanales dataset, both genes are present in nine copies, while one copy contains a fusion of the genes. In the BUSCO Fabales dataset, only the methyltransferase gene is present and one of the copies contains the artificial fusion of the other gene. In the BUSCO Poales dataset, six of the ten gene copies contain the artificial fusion of the two genes, the other four genes are copies of the methyltransferase.
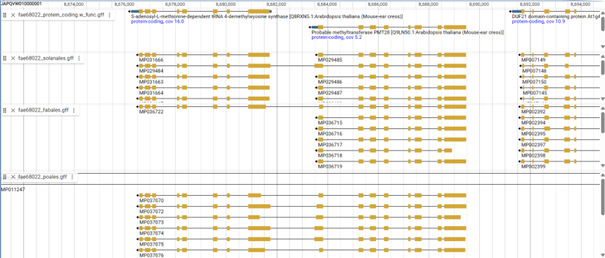
How does this affect the completeness check of the annotation? When checking BUSCO completeness with the Solanales dataset, the tRNA-4-demethylwyosine synthase is classified as ‘missing’ both in the genome assembly check and in the genome annotation, the methyltransferase is classified as ‘single, complete’. When checked with the Fabales dataset, the tRNA-4-demethylwyosine synthase does not exist and the methyltransferase is classified as ‘single, complete’. When checked with the Poales dataset, the tRNA-4-demethylwyosine synthase and the methyltransferase are categorised as ‘duplicate’.

Although the tRNA-4 demethylwyosine synthase is mapped to the genome in the Solanales dataset (see genome browser screenshot), it is classified as ‘missing’. The only explanation for this strange categorisation is that the methyltransferase gets a higher score, the single artificial fusion gene belongs to the methyltransferase gene group, therefore the tRNA-4 demethylwyosine synthase hits are removed as overlapping, and consequently the tRNA-4 demethylwyosine synthase is ‘missing’ because no hit remains. When checked with the Poales dataset, both genes are classified as ‘duplicates’ because they both correspond to the same artificial fusion gene.
Conclusions
This is not a single outlier. There are many other cases of artificially fused genes in the BUSCO datasets that lead to the same proven effect. In the case of these artificially fused genes, the result of the BUSCO completeness analyses depends on the corresponding genome assembly. If the genes that are part of the artificial fusion are not in close proximity in the genome assembly, the completeness check will give a different result.