Functional annotation is the process of assigning biological meaning to identified gene sequences. It involves predicting the roles of genes, transcripts, and proteins by comparing them to known databases, identifying protein-coding regions, functional domains, motifs, and gene ontology (GO) terms. Functional annotation also includes the identification of regulatory elements, pathways, and interactions, providing insights into the biological processes, molecular functions, and cellular components associated with each gene.
In Part 1 and 2, you were given an overview of the problem of assigning gene/protein names based on known databases, sequence databases and protein family databases such as PANTHER. In this part, we will take a look at what the databases included in the InterProScan package cover.
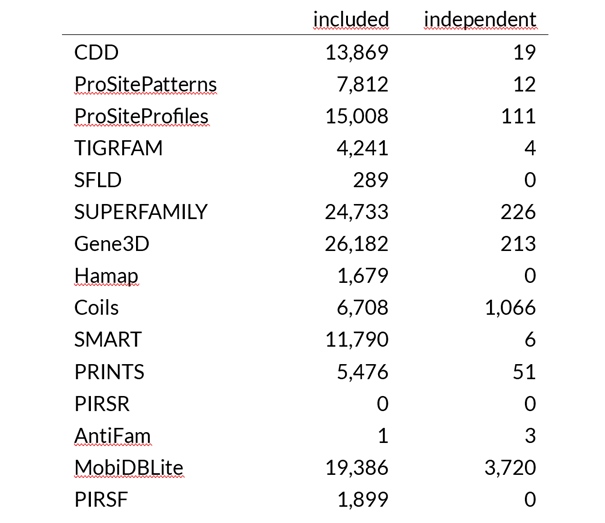
For demonstration purposes, I selected a dataset containing 46,664 protein sequences from a genome annotation. A genome annotation should cover everything an organism needs to live and represent a typical use case without certain protein families being overrepresented. Of the proteins from the genome annotation, 32,945 received a Pfam domain assignment and 38,141 received a PANTHER assignment. These figures already indicate a large overlap between the assignments. What about the remaining 15 tools included in InterProScan? The following table shows that only a few assignments from these tools are not already included in Pfam or PANTHER assignment. For example, the domain architecture assignment tools CDD and SMART contain far fewer reference domains in their databases, and only a few of their domains are not included in Pfam or have slightly different domain sizes, causing their matches to exceed the cutoff. Predictions of coiled-coils using COILS have proven to be random (see one of the previous posts). MobiDBLite is used to predict intrinsically disordered regions. Given the predictions for the example dataset, this would mean that 50% of all proteins and those with Pfam and/or PANTHER assignments contain disordered regions, which seems highly unlikely given the many globular enzymes and structural proteins.
Table: InterProScan analysis of a protein sequence dataset. The tools with most hits (Pfam and PANTHER) were regarded as reference. Search hits with other tools are given as included in the subset of proteins with Pfam and PANTHER hits or independent (not included), respectively. Software and version: InterProScan 5.72-103.0. Total proteins: 46,664. Thereof 32,945 proteins with Pfam hits and 38,141 proteins with PANTHER hits.
Conclusion
When Pfam is used within InterProScan, the inclusion of additional databases/tools does not provide any significant additional information. PANTHER should be excluded because it contains too many ambiguous, non-informative and false positive (e.g. transposon) protein families. COILS should be avoided entirely due to its random prediction quality. MobiDBLite has not yet been evaluated in detail, but should be avoided as it appears to strongly overestimate intrinsically disordered protein regions.