Functional annotation refers to the process of assigning biological significance to identified gene sequences. This involves inferring the functions of genes, transcripts, and proteins by aligning them with established databases, and detecting features such as protein-coding regions, functional domains, motifs, and gene ontology (GO) terms. It also encompasses the identification of regulatory elements, biological pathways, and molecular interactions, offering insights into the roles each gene plays in biological processes, molecular functions, and cellular components.
In Part 1, you were given an overview of the problem of assigning gene/protein names based on known databases. Here we will take a look at one of the databases used for assigning functions to gene/protein sequences, namely PANTHER. The PANTHER knowledgebase aims to support biomedical and related research by offering comprehensive data on the evolution of protein-coding gene families, with a particular focus on protein phylogeny, functional annotation, and genetic variations that influence protein function.
The latest release of PANTHER includes 15,683 gene families and approximately 128,000 subfamilies. Each family is assigned a name, often derived from one of the earliest identified members found in databases such as UniProt, GenBank, or other major sequence repositories. As a result, the gene or protein names listed in these sources have become the family names within PANTHER. Consequently, about one-third of these names include vague or non-informative prefixes and suffixes like 'putative', 'related', 'like', or 'hypothetical'. A small percentage of families or subfamilies lack a name entirely or are simply labeled as '(sub)family not named'. Since genome annotations rely heavily on existing database entries, these ambiguous names are transferred to newly annotated genomes, often with additional layers of non-informative modifiers, compounding the ambiguity. This practice has occasionally led to amusing and unintended naming chains.
1,235 families have the term 'putative-related' in their name, and 13 families have the term 'putative-related-related' in their name. One family is even named 'putative-related-related' without any additional description, another family is named 'protein, putative-related-related'.
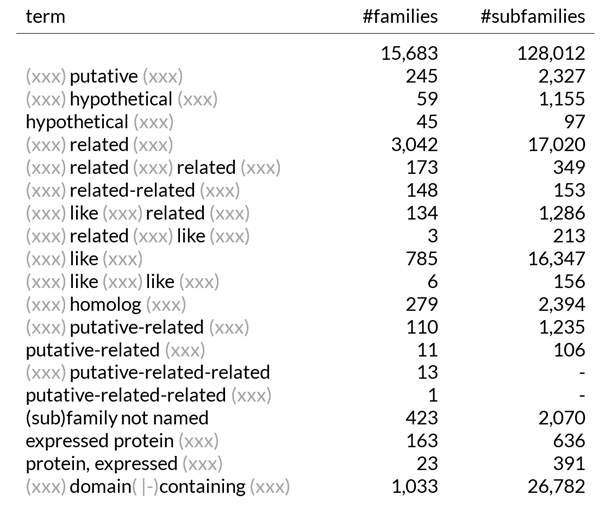
Of particular importance in this naming are descriptive additions that have a biological meaning, such as ‘expressed’ or ‘domain-containing’. If non-annotated proteins receive such family assignments in the process of functional annotation, the name of the newly annotated protein implies that this protein contains the assigned domain or is expressed. This is of course not the case for all annotated proteins and leads to misleading assumptions and experiments and could also add to the confusion in the public databases if the annotation is submitted/included. If the annotation is included, further error propagation occurs. Another major problem with PANTHER is that the definition and labelling of the families started many years ago when only a few genome annotation projects were available. Accordingly, many families contain the technical gene ID of the gene/protein of that annotation, which is not informative, and many of the families represent non-genetic sequences from incorrect gene predictions such as transposon fragments and other repeat regions. The problem of passing on names containing biological terms such as ‘domain-containing’ will be addressed in one of the next newsletters.
Conclusion
When you perform functional genome annotation, you must carefully analyse, rename and reassign all mappings from automated processes such as InterProScan or BLAST2GO that assign functions from third-party databases such as PANTHER or third-party software such as COILS. If you don't do this renaming and remapping, each of your functional annotations will contain and propagate these mismappings. At GOENOMICS, we have written hundreds of lines of code to carefully analyse these mappings and avoid misleading functional annotations.